
RETINOBLASTOMA
FAQ
Retinoblastoma is the most common intraocular malignancy in children. Approximately 60% of cases are unilateral (involving one eye), while the remaining 40% are bilateral (both eyes). Patients diagnosed with this malignancy are categorized whether the gene mutation is germline (from sex cells: eggs and sperm) or somatic (from body cells), as this has a bearing on the clinical management.
The retinoblastoma susceptibility gene RB1 is a tumor-suppressor gene. Two active copies of the RB1 gene are normally found in human cells. Both copies must be mutated (“two-hit mechanism”) to lead to retinoblastoma tumor formation. The initial mutation, which may occur in either germline or somatic cells, inactivates one copy of the gene. The second mutation inactivates the other copy in somatic cells.
If the first mutation occurs in germline cells (seen in 30% of cases), the patient has heritable retinoblastoma. Patients with germline RB usually have bilateral and multifocal tumors. They also have a significantly increased risk for developing secondary tumors in the brain (primitive neuroectodermal tumor or PNET). Furthermore, children will have an increased risk of developing retinoblastoma, as this trait is transmitted in an autosomal dominant fashion with high penetrance (90%).
If the first mutation occurs in a somatic cell of the developing retina, it is not heritable.
Genetic counseling for families with known retinoblastoma can help to determine whether other family members are at risk for developing the disease.
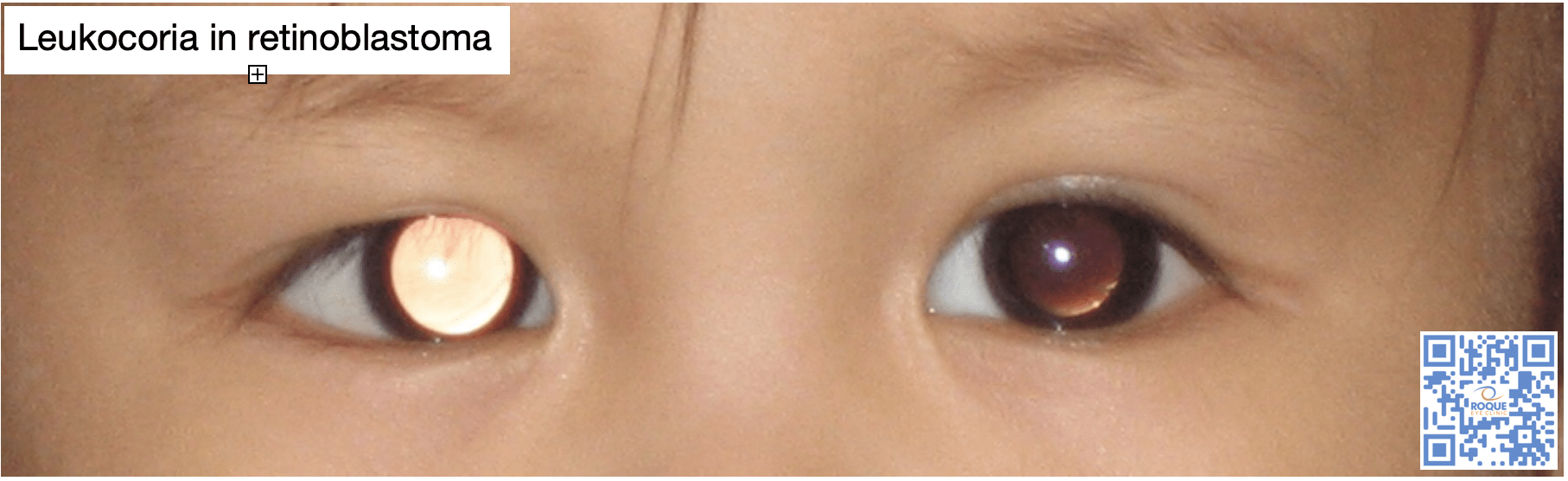
Leukocoria (whitening of the red reflex) is the most common presentation of retinoblastoma, and all infants or children with an abnormal red reflex require immediate referral to a pediatric ophthalmologist or retina specialist. Offsprings and siblings of patients with retinoblastoma need to undergo newborn screening and regular screenings during childhood.
Eye examination starts with the age-appropriate visual acuity testing, pupillary reflex testing, and slit lamp examination. The dilated eye examination is the last and most important part of the eye examination. If there is any suspicion for retinoblastoma, the patient should undergo an examination under anesthesia. An eye examination under anesthesia with careful scleral depression will not only confirm the diagnosis, but it will also determine the exact location as well as the extent of the tumor(s) and the tumor staging. This is a good time to do photodocumentation for future reference.
Other diagnostics tests are necessary. A and B scan ultrasound will help define tumor height and thickness, as well as confirm the presence of calcifications and associated retinal detachment and calcification. Cranial and orbital magnetic resonance imaging (MRI) is often performed to evaluate optic nerve involvement, extraocular extension, and also to rule out the possibility of concomitant primitive neuroectodermal tumor (PNET). MRI is currently the preferred imaging modality because of the radiation associated with CT scans.
Bone marrow examination or lumbar puncture may also be performed in patients with extraocular extension to rule out cerebrospinal fluid (CSF) or bone marrow metastases.
The current classification was developed to better predict those with intraocular RB who are likely to be cured without the need for enucleation or external-beam radiation treatment:
- Group A tumor size <3mm, away from fovea and disc
- Group B tumor size >3mm, macular or juxtapapillary, or with subretinal fluid
- Group C tumor plus focal subretinal or vitreous seeding within 3 mm of tumor
- Group D tumor plus diffuse subretinal or vitreous seeding >3mm from tumor
- Group E extensive tumor >50% of globe with or without neovascular glaucoma, hemorrhage, extension of tumor to optic nerve or anterior chamber
The treatment of retinoblastoma is aimed at preserving life, preserving the globe, and preserving vision, in that specific order, with the minimum side effects and complications.
Enucleation remains the definitive treatment of intraocular retinoblastoma (in Group E), particularly in the majority of patients who present with unilateral disease.
Treatment modalities that may be successful in preserving the globe include combined systemic chemotherapy with focal consolidation (also called chemoreduction), intra-arterial chemotherapy, and for small tumors, focally destructive therapy (cryopexy, laser photocoagulation, hyperthermia and plaque irradiation).
Radiation therapy is avoided, if possible, but still has a role to preserve vision in recurrent tumors.
Chemoreduction is a medical approach to shrink the tumor by combining the effects of systemic chemotherapy and focal consolidative procedures. Shrinking the tumor increases the success of focal therapies. Focal therapy directly destructs the tumor cells and also breaks down the blood ocular barrier and increases penetration of chemotherapeutic agents into the eye. This combination is one of the main globe-salvaging options in retinoblastoma management.
Current studies are now investigating the effectivity of more localized delivery of chemotherapeutic agents, in combination with chemoreduction techniques. This will help decrease the systemic side effects of chemotherapeutic agents. Recently, significant attention has been focused on intra-arterial delivery of chemotherapeutic agents either via the carotid artery or ophthalmic artery in unilateral retinoblastoma. Investigators have yet to determine the safe dosage of the best chemotherapeutic agent for this approach.
Eyes in Group E and eyes that showed progression despite conservative treatments eventually require enucleation. To make sure that the cut end of the nerve is tumor free, enucleation requires removal of as much optic nerve as possible. At the time of enucleation, the largest orbital implant is placed in order to encourage normal development of the pediatric orbit. The globe is then sent for pathologic evaluation.
Pathologic evaluation determines whether there are any risk factors for extraocular spread. Pathologic risk factors (PRF) include: 1. choroidal invasion, 2. post-laminar invasion of the optic nerve, 3. scleral invasion, and 4. involvement of the anterior chamber. When PRFs are present, adjuvant chemotherapy should be considered considered to decrease the risk of extraocular relapse.
Patients who have undergone enucleation should be followed to ensure there is no evidence of tumor in the other eye and will also need fitting for an ocular prosthesis.
For extraocular retinoblastoma limited to the orbit, neoadjuvant chemotherapy is started to shrink the tumor. This is then followed by surgical debulking and post-operative chemotherapy, and radiation if necessary.
For those with systemic metastases, especially those with CNS involvement, aggressive treatment with high dose chemotherapy (HDC) and autologous stem cell rescue (ASCR) is recommended. HDC involves the administration of high doses of chemotherapeutic agents with the aim to overcome tumor resistance and completely eradicate neoplastic cells. Unfortunately, these lethal doses are also myeloablative and concurrent ASCR must be performed to allow future regeneration of the bone marrow. This is also the approach for those with concurrent intracranial tumor (“trilateral retinoblastoma”).
Patients who undergo any form of globe-sparing treatment need frequent follow-up eye examinations. Tumor regression is checked closely, documenting the appearance, size, location and number of tumors during each examination. Regressed tumor appears like a white calcific mass or a translucent fish flesh. Eye examinations under general anesthesia is ideally done every 6-8 weeks until age three, and followed less frequently if the condition becomes quiescent.
Patients with the hereditary mutation are at increased risk of developing secondary malignancies throughout the body for the rest of their lives. Patients who have receive radiation are at higher risk for secondary tumors in the field of treatment. It is mandatory for all retinoblastoma patients to undergo long-term follow-up, with special vigilance if patients have germline mutations.
- Dimaras, H., Corson, T., Cobrinik, T., White, A., Zhao, J., Munier, F., Abramson, D., Shields, C., Chantada, G., Njuguna, F., Gallie, B. (2015). Retinoblastoma. Nat Rev Dis Primers, 1:15021.
- Peckacka, A. (2020). The role of intra-arterial chemotheraphy in the management of retinoblastoma. J Ophthalmol, 2020: 3638410.
- Wyse, E., Handa, J, Friedman, A., and Pearl, M. (2016). A review of literature for intra-arterial chemotherapy used to treat retinoblastoma. Pediatr Radiol, 46(9): 1223-1233.

BOOK AN APPOINTMENT
It takes less than 5 minutes to complete your online booking. Alternatively, you may call our BGC Clinic, or our Alabang Clinic for assistance.
OUR SPECIALIST

DR. BARBARA ROQUE
MD, DPBO, FPAO, FPCS
Dr. Barbara Roque is a specialist in pediatric ophthalmology, adult strabismus, and ophthalmic genetics. Her private practice began in 2006, after her post-graduate fellowship training at The Children’s Hospital in Westmead, University of Sydney System, Australia. Her patients are mostly children with ocular disease, refractive errors, cataracts, and eye misalignment.
OUR CLINICS
BGC CLINIC
- ST. LUKE'S MEDICAL CENTER GLOBAL CITY
2/F Medical Arts Building 217
Rizal Drive corner 5th Avenue
Bonifacio Global City, Taguig 1634
Philippines
SLMC CLINIC HOURS
- 9am - 12pm
Appointments only
ALABANG CLINIC
- ASIAN HOSPITAL AND MEDICAL CENTER
5/F Medical Office Building 509
2205 Civic Drive, Filinvest City
Alabang, Muntinlupa 1781
Philippines
AHMC CLINIC HOURS
- 1pm - 4pm
Appointments only
BOOK AN APPOINTMENT
It takes less than 5 minutes to complete your online booking. Alternatively, you may call our BGC Clinic, or our Alabang Clinic for assistance.



{kind=link}
{kind=link}
{kind=link}