
ANTERIOR SEGMENT DYSGENESIS
FAQ
Anterior segment dysgenesis (ASD) is a congenital malformation of the front part of the eye. It is a spectrum of disorders that affect the development of the frontal parts of the eye which includes the cornea, the iris, the ciliary body and the lens.
- Underdevelopment of the iris (iris hypoplasia)
- An enlarged or reduced corneal size diameter
- Growth of new blood vessels (vascularization) and opacity of the cornea (leukoma)
- Thickened and displaced Schwalbe’s line (posterior embryotoxon)
- Displacement of the pupil (corectopia)
- More than one pupillary opening (polycoria)
- Abnormal iridocorneal angle
- Displacement fo the lens (ectopia lentis)
- Absent lens (aphakia)
- Cataracts
- The iris adheres to the cornea (anterior synechia)
- Thinning of the cornea (posterior keratoconus)
The subtype of ASD depends on the combination of specific abnormalities mentioned above and the genetic cause. Some may even be associated with neurologic abnormalities. Glaucoma develops in approximately 50% of cases during infancy or much later.
This is composed of two anomalies: Axenfeld anomaly (posterior embryotoxon) and Rieger anomaly (iris stromal atrophy in the form of holes or pseudo-holes and corectopia).
This is a multisystem condition composed of the above plus accompanying facial, dental, umbilical, and skeletal abnormalities . The ocular findings are extremely variable between and within families. The typical facial features include broad nasal root with telecanthus, maxillary hypoplasia, and a prominent lower lip. The dental anomalies include hypodontia and partial anodontia; the remaining teeth have small crowns. The failure of involuntion of the periumbilical skin is a cardinal feature of this syndrome. Other rarely associated abnormalities include: anal stenosis, hypospadia, empty or enlarged sella turcica, growth hormone deficiency, psychomotor retardation, mild deafness, dilatation of the cerebral ventricles, and congenital heart disease.
Peters anomaly may be isolated or may be associated with ocular defects such as microcornea, anterior polar cataracts, glaucoma with or without buphthalmos, spontaneous corneal perforation, aniridia, congenital aphakia, peristence of the hyaloid system and total posterior coloboma of the retina and choroid. Associated systemic congenital anomalies include short stature, abnormal ears, cleft lip/palate abnormalities, malformation of the genitourinary system, cardiovascular anomalies, defects of the extremities and mental retardation.
Anterior segment dysgenesis is due to an abnormality in the development of neural crest cells (termed neurocristopathy).
Axenfeld-Rieger syndrome is associated with PITX2 and FOXC1 genes and inherited as an autosomal dominant manner. It is also associated with chromosomal aberrations in the following chromosomes: 4, 6, 10, 13, 16, and 22. Males and females are equally affected. It is fully penetrant and with variable expressivity.
Rieger syndrome is associated with RIEG1 RIEG2 RIEG3 genes. Males and females are equally affected. Like Axenfeld-Rieger syndrome, it is fully penetrant and with variable expressivity.
Compared to the first two forms of ASD, Peters’ anomaly is not monogenic. It has been documented to be associated with mutations in PAX6, PITX2, CYP1B1, and FOXC1. Familial isolated Peters anomaly is recessively inherited in majority cases but there are occasional pedigrees with autosomal dominant transmission. Peters anomaly with multiple congenital malformations has been reported in patients with chromosomal abnormalities in 11, 4, 18, 21 and 2.
A comprehensive eye examination is vital to the diagnosis and management of anterior segment dysgenesis. Young children should be considered for an eye examination under general anesthesia. Visual field monitoring should be started as soon as the child is able. All family members should be examined.
Ultrasound biomicroscopy (UBM), optical coherence tomography (OCT) and B scan ultrasonography (B scan UTZ) are critical to surgical planning. In Peters’ anomaly, the visual prognosis depends on the severity of corneal opacification and associated ocular malformations. A combination of penetrating keratoplasty and cataract extraction improves the visual prognosis. Histopathologic exam at the time of corneal grafting could confirm the diagnosis.
Affected siblings and children of patients with ASD should be monitored for the development of glaucoma before the development of visual field loss.
Genetic counseling should take into consideration the autosomal dominant mode of inheritance of the disease, the nearly complete penetrance of the gene and the variability of clinical presentation. Chromosomal studies should be obtained in ASD patients with multiple congenital malformations.
Deep amblyopia may be difficult to treat, particularly in unilateral Peters’ anomaly with severe corneal opacification and cataract. Post-surgery glaucoma may develop in approximately 50%, and could lead to vision loss if not detected.
- >
- Chen, W., Xiang, D., and Hu, L. (2020). Ultrasound biomicroscopy detects Peters’ anomaly and Rieger’s anomaly in infants. J Ophthalmol, Mar 23, 2020.
- Chrystal, P., and Walter, M. (2019). Aniridia and Axenfeld-Rieger Syndrome: Clinial presentations and molecular genetics and current/emerging therapies. Exp Eye Res. 189: 107815.
- Gauthier, A, and Wiggs, J. (2020). Childhood glaucome genes and phenotypes: Focus on FOXC1 mutations causing anterior segment dysgenesis and hearing loss. Exp Eye Res.

BOOK AN APPOINTMENT
It takes less than 5 minutes to complete your online booking. Alternatively, you may call our BGC Clinic, or our Alabang Clinic for assistance.
OUR SPECIALIST

DR. BARBARA ROQUE
MD, DPBO, FPAO, FPCS
Dr. Barbara Roque is a specialist in pediatric ophthalmology, adult strabismus, and ophthalmic genetics. Her private practice began in 2006, after her post-graduate fellowship training at The Children’s Hospital in Westmead, University of Sydney System, Australia. Her patients are mostly children with ocular disease, refractive errors, cataracts, and eye misalignment.
OUR CLINICS
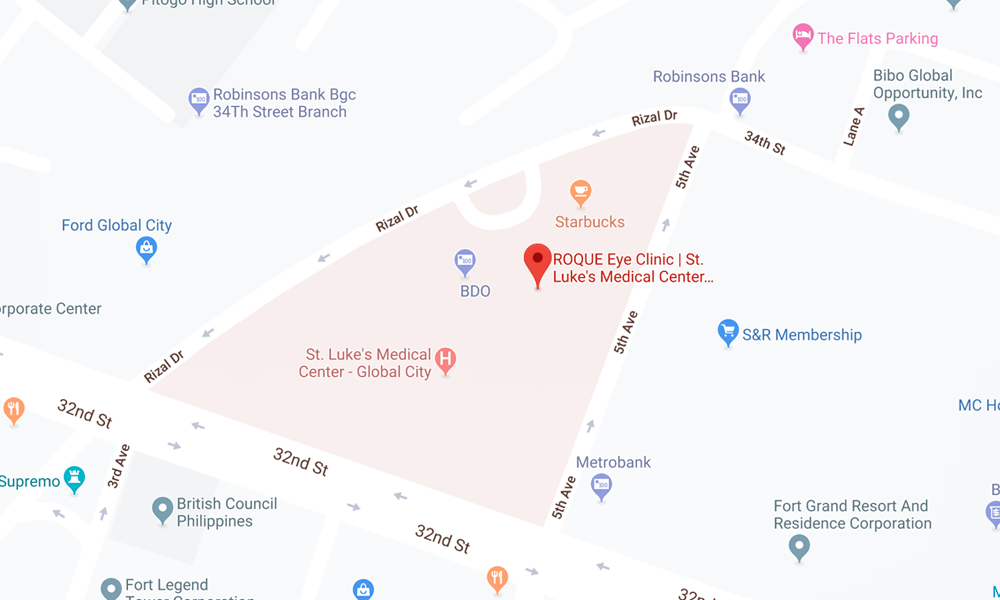
BGC CLINIC
- ST. LUKE'S MEDICAL CENTER GLOBAL CITY
2/F Medical Arts Building 217
Rizal Drive corner 5th Avenue
Bonifacio Global City, Taguig 1634
Philippines
SLMC CLINIC HOURS
- 9am - 12pm
Appointments only
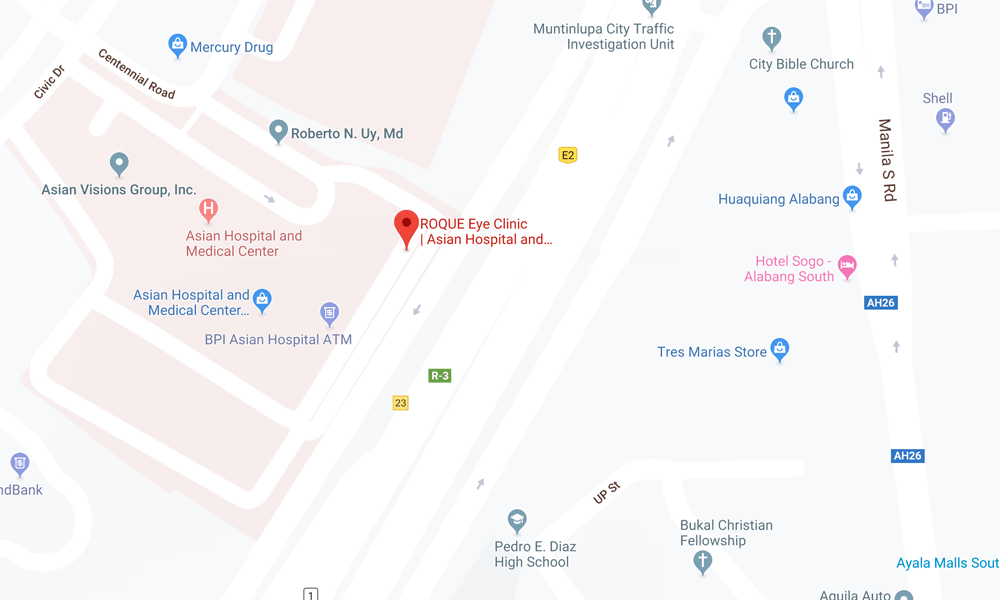
ALABANG CLINIC
- ASIAN HOSPITAL AND MEDICAL CENTER
5/F Medical Office Building 509
2205 Civic Drive, Filinvest City
Alabang, Muntinlupa 1781
Philippines
AHMC CLINIC HOURS
- 1pm - 4pm
Appointments only
BOOK AN APPOINTMENT
It takes less than 5 minutes to complete your online booking. Alternatively, you may call our BGC Clinic, or our Alabang Clinic for assistance.



{kind=link}
{kind=link}
{kind=link}